
Quantum Espresso Convergence Diary ($Na_{3}Co_{2}SbO_{6}$)
I face the problem of bad convergence when doing SCF calculation. To solve the problem, keeping documentary is a good idea …
1st Version
Directly tried the SOC with zigzag antimagnetic order, plus LDA+U as below:
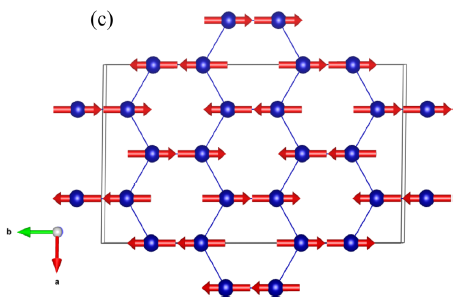
And the pw.x input file below (the input file is done according to Wien2K results):
1 | &CONTROL |
And the outcome is just not converging. In the first iteration scf accuracy < 3000Ry. So bad! Besides, the speed is very very slow (1.4h for each iteration).
2nd Version
Maybe it’s not wise to try such complexity at the beginning. So I cut down k-mesh, ecutwfc, and ecutrho (after referring paper).
1 | ! calculation = 'vc-relax' , |
It’s still don’t converge… Still so slow…(about 1.2h for each iteration)
Note that the first iteration scf accuracy still < 3000 Ry, I think maybe something is wrong with my structure settings. So I turn off the SOC, LDA+U and magnetic order settings, just calculate the unit cell of normal $Na_{3}Co_{2}SbO_{6}$.
3rd Version
This time, just calculate the nonmagnetic system.
input file :
1 | ! calculation = 'vc-relax' , |
The calculation is even more weird…
First iteration scf accuracy is about 2~3 Ry, but as iteration goes on, the accuracy is firstly decreasing, and then continuously going up, to 500 Ry finally. And I just don’t know why.
But have to confess that the calculation this time is raised a lot (about 5min for each iteration).
4th Version
Searching the Internet, I found several useful advice for better convergence from here. Listed them below.
Lowering mixing_beta and increasing mixing_ndim.
mixing_beta : 0.7 default, but try 0.1~0.3 or even smaller 0.05 for very long slab
mixing_ndim : 8 default, but try 12
Raise ecutwfc and ecutrho.
In this way, make changes on the basis of above input file:
1 | ecutwfc = 90.0 , |
1 | mixing_beta = 0.5 , |
And the outcome is still weird.
After the beginning reducing of scf accuracy, the calculation still failed to converge.
The scf accuracy jumped between 0.002 and 0.00002 at last. There is still someing to do …
5th Version
Searching the Internet, I find one possible solution here.
Where one of the answer remind me of changing occupation, i.e. :
1 | ! occupations = 'tetrahedra_opt', |
At last, it converged! So I calculated DOS.
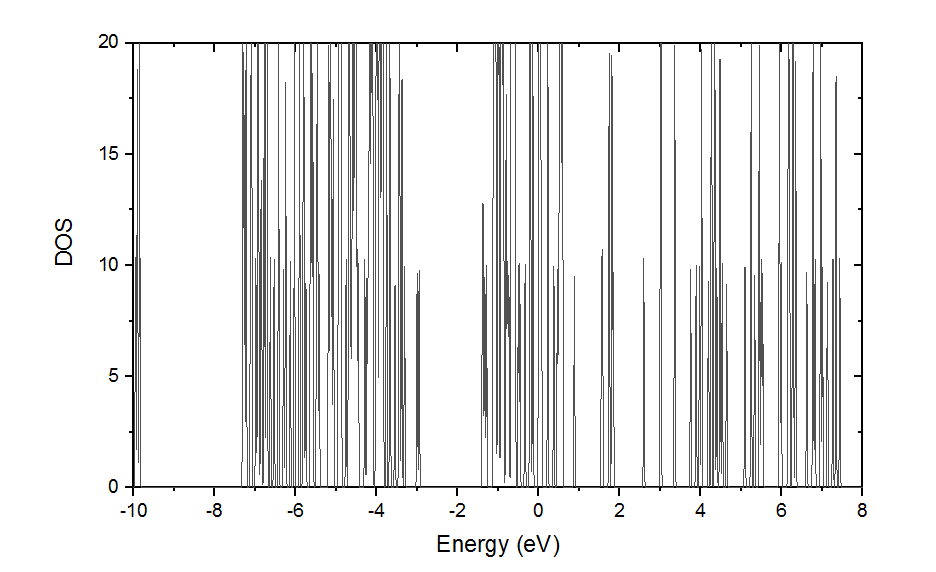
Comb-like sharp peaks appeared. This reminded me previous conditions that these peaks showed up when I do scf calculation using ‘occupation = tetrahedron’ instead of ‘occupation = tetrahedra_opt’. This means though ‘occupation = smearing’ may be a possible way to get convergence, I still need to try with ‘tetrahedra_opt’ .
6th Version
So go back to 4th Version.
First, try to run input file of 4th Version with ‘occupation = tetrahedra_opt’, the calculation stopped after 130 iterations, failing to get convergence. But the scf accuracy once reached 0.00002, close to conv_thr. Is it possible to make changes on the basis of it?
So I increased ecutwfc, ecutrho and reduced the mixing_beta on the basis of 4th Version code.
1 | ecutwfc = 100.0 , |
1 | mixing_beta = 0.3 , |
1 | nbnd = 120 , |
Still don’t converge!
7th Version
Things changed when I use different pseudo-potential. The 1st-6th Version used PBE pseudopotential generated from pslibrary1.0.0. But the converging performance is bad somehow. So I used pseudopotential from SSSP pseudopotential. Then it began to converge. First, try $Na_{3}Co_{2}SbO_{6} $with nonmagnetic and non-LDA+U and nonSOC case. See the pw.x input file below.
1 | &CONTROL |
The selection of pseudopotential is really important to calculation results!
Actually there is another reason(but a minor factor, without changing pseudopotential, it still won’t get final convergence). The server contains 16 cores for us(me and another guy) to calculate. Though we agree that each of us use 8 cores, we actually use the same 8 cares! And the speed is extremely slow. After using the command below:
mpirun -np 8 --cpu-set 8,9,10,11,12,13,14,15 pw.x < xx.scf.in > xx.scf.out
or
mpirun -np 8 --cpu-set 0,1,2,3,4,5,6,7 pw.x < xx.scf.in > xx.scf.out
I can directly select specific cores to work for me. Thus, the calculation this time is much faster.
Then calculate its total dos and pdos of the system. Below is the result.
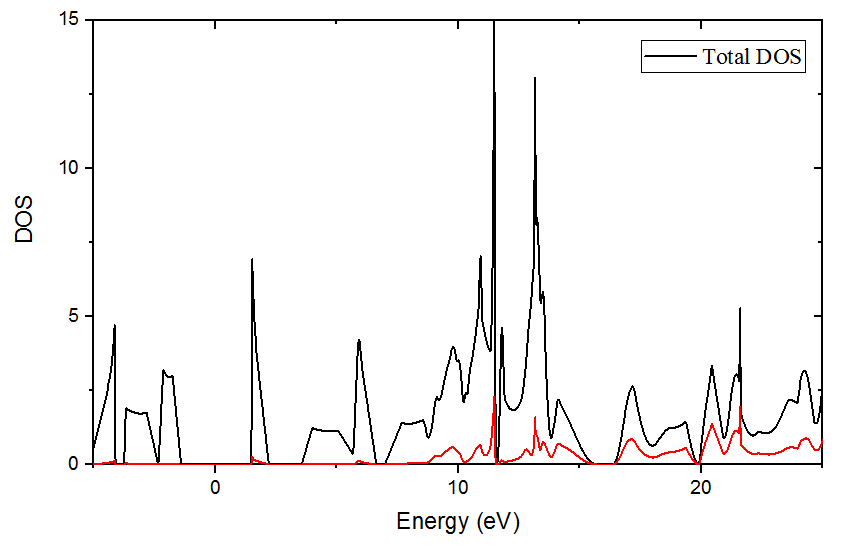
The comb-like sharp peaks also disappear!
Then we can begin calculation with zigzag magnetization, SOC and LDA+U.
8th Version
The input file of pw.x for NCSO with zigzag magnetization, SOC and LDA+U is below.
1 | &CONTROL |
With this, we can calculate the electronic properties of this system.
The dos and pdos result are below.
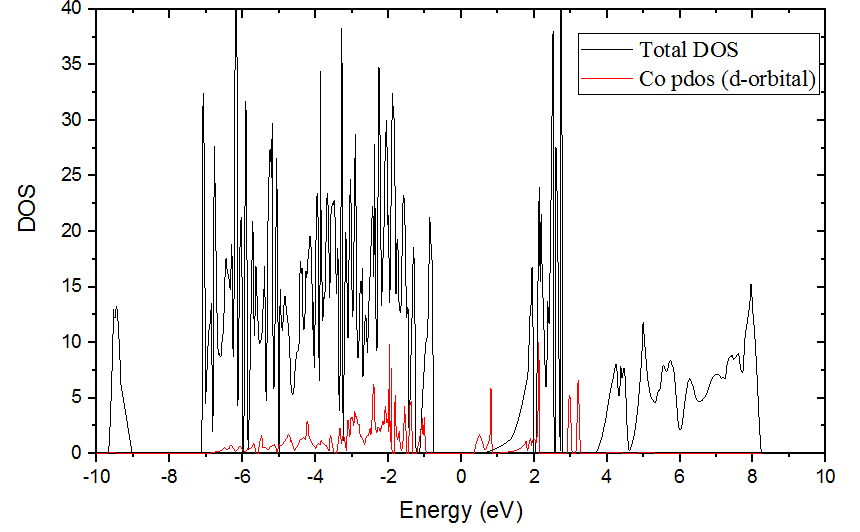
Looks like good!
Note that the pdos and dos aren’t completely consistent with each other. I guess that the dos and pdos are two separate calculation process, and some little errors between them are generated from this.
Summary
Listed below are some possible ways to achieve convergence.
- Lower the number of K points.
- Decrease the number of mixing_beta(0.7 -> 0.1~0.3 or even 0.05).
- Change pseudopotential.
- Optimize your structure.
- Increase your ecutwfc and ecutrho.
The wisest way when faced with a new calculation, try with simplest form first, and then step to the final result.
Don’t worry! Be happy!